Microbial RNA-seq#
Summary of methods#
- Index generated with RSEM
- Filter and trim reads with fastp
- Strand determination with how are we stranded here
- Quality checking of reads with FastQC
- Reads mapped to reference and quantified with RSEM
- Alignment post-processing and QC with Picard
- Aggregation of QC tables using MultiQC
Parameters#
Required parameters:#
-
--pubdir- Default:
/<PATH> - Description: The directory that the saved outputs will be stored.
- Default:
-
-w- Default:
/<PATH> - The directory that all intermediary files and nextflow processes utilize. This directory can become quite large. This should be a location on scratch space or other directory with ample storage.
- Default:
-
--sample_folder- Default:
/<PATH> - The path to the folder that contains all the samples to be run by the pipeline. The files in this path can also be symbolic links.
- Default:
-
--fasta- Default:
/<PATH> - Path to the reference genome in FASTA format
- Default:
-
--gff- Default:
/<PATH> - Path to the annotation for the reference genome in GFF3 format
- Default:
-
--read_type- Default:
PE - Comment: Type of reads: paired end (PE) or single end (SE).
- Default:
Optional parameters:#
General optional parameters:#
-
--strandedness- Default:
NA - Supported options are
reverse_stranded,forward_stranded,non_stranded - Strandedness of libraries is determined automatically by how are we stranded here, and this parameter is used as a fallback if the automatic strand determination fails
- Default:
-
--concat_lanes- Default:
false - Options:
falseandtrue. Default:false. If this boolean is specified, FASTQ files will be concatenated by sample. Used in cases where samples are divided across individual sequencing lanes.
- Default:
-
--rsem_index- Default:
/<PATH> - Enables the use of indices that were previously generated with this pipeline, by providing the path to that directory. Will generate an error if any files are missing. If using this parameter, do not use
--fastaand--gff
- Default:
-
--extension- Default:
.fastq.gz - Expected file extension for input read files, modify if files are not compressed or have a different form (e.g. ".fastq" or ".fq.gz")
- Default:
-
--pattern- Default:
*_R{1,2}* - Expected R1/R2 matching pattern for paired-end read files in the
--sample_folderpath. In concert with--extensionthe default values will match files such assampleID.L001.R1.fastq.gzandsampleID.L001.R2.fastq.gz
- Default:
-
keep_intermediate- Default:
false - If
trueworkflow will output intermediate alignment files (unsorted BAMs, etc.)
- Default:
-
keep_reference- Default:
false - If true workflow will save a copy of the RSEM indices to the output directory
- Default:
fastp filtering parameters:#
-
--quality_phred- Default: 15
- Quality score threshold.
-
--unqualified_perc- Default: 40
- Percent threshold of unqualified bases to pass reads.
bowtie2 parameters:#
--seed_length- Default: 25
- From RSEM manual: If RSEM runs Bowtie, it uses this value for Bowtie's seed length parameter. Any read with its or at least one of its mates' (for paired-end reads) length less than this value will be ignored.
Pipeline Default Outputs#
| Naming Convention | Description |
|---|---|
rsem.merged.gene_counts.tsv |
RSEM-generated gene-level raw counts merged across all samples |
rsem.merged.gene_tpm.tsv |
RSEM-generated gene-level TPM counts merged across all samples |
rsem.merged.isoform_counts.tsv |
RSEM-generated isoform-level raw counts merged across all samples |
rsem.merged.isoform_tpm.tsv |
RSEM-generated isoform-level TPM counts merged across all samples |
rnaseq_report.html |
Nextflow autogenerated report |
index/ |
RSEM-generated indices saved with optional parameter keep_reference |
multiqc/ |
MultiQC report summarizing quality metrics across all samples in the run |
${sampleID}/bam/ |
RSEM-generated alignments of reads to the reference genome and transcriptome |
${sampleID}/stat/ |
QC, strand, and Picard metrics files for each sample |
${sampleID}/${sampleID}.genes.results |
RSEM-generated quantification of gene-level count abundances |
${sampleID}/${sampleID}.isoforms.results |
RSEM-generated quantification of transcript-level count abundances |
trace.txt |
Nextflow trace of processes |
Workflow Validation#
The genome assembly and annotation for Enterococcus faecalis V583 were
downloaded from NCBI
and used to generate simulated RNA-seq reads with the function
simReads in the R package Rsubread. To test the performance of the workflow, we generated three sets of
"control" paired-end reads, in which the TPM values were assigned based on
random sampling, and three sets of "case" control reads, in which 50 genes
were randomly selected to have substantially higher TPM values.
We then ran the workflow using the E. faecalis reference to generate indices, and compared the raw count values quantified by RSEM with the "true" count values expected from the simulated reads generated by Rsubread.
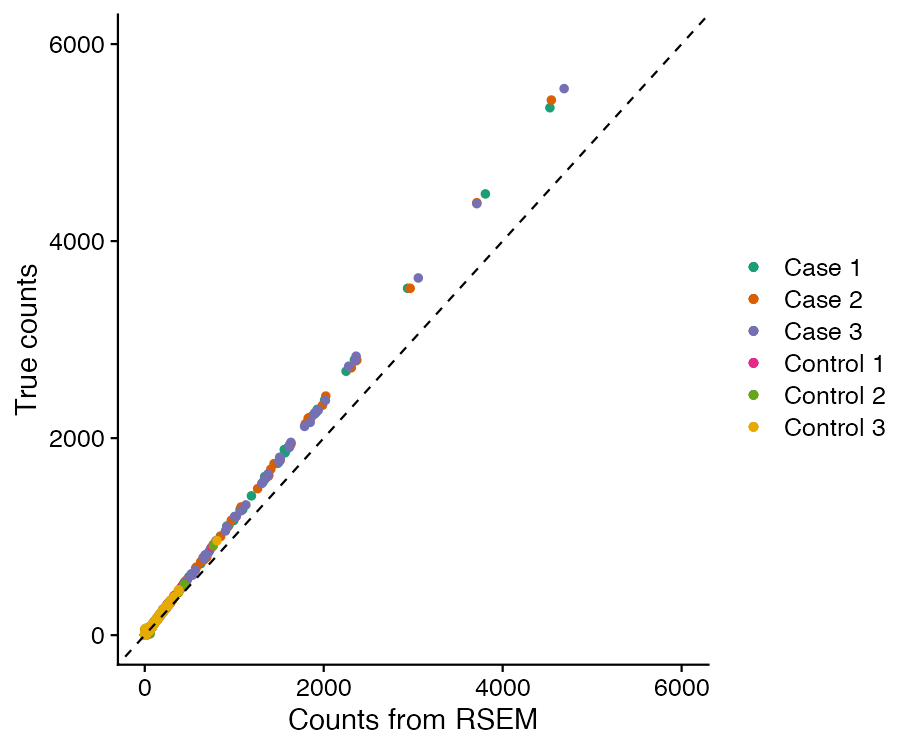
The dashed line represents a 1:1 reference, and the deviation of the counts quantified by RSEM from the "True" counts is due to the quality filtering of the read data, as the reads were simulated to include sequencing error.
We then performed differential expression analyses on both the true counts and the counts estimated by RSEM in the workflow.
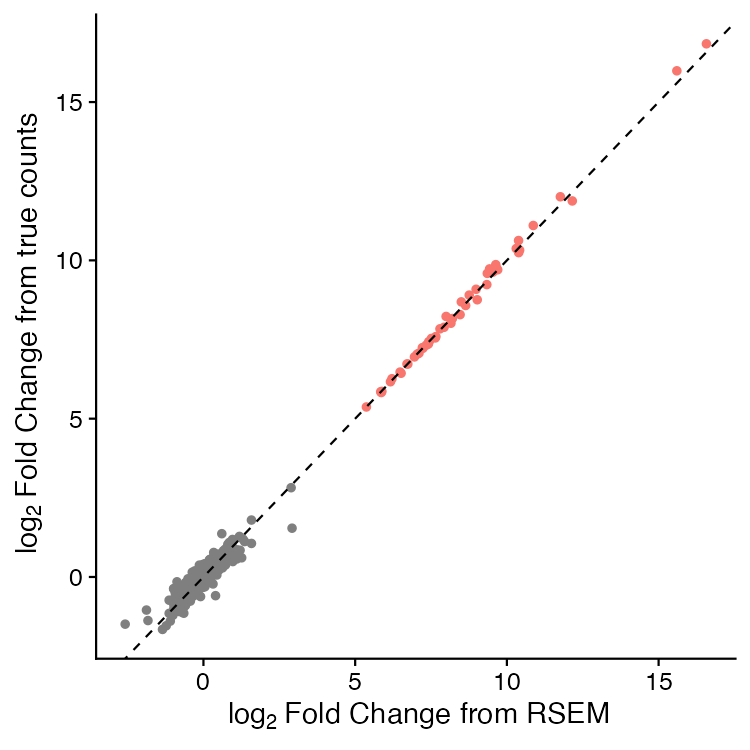
Again, the dashed line is a 1:1 reference. The red points are those which would be scored as "significant" with standard thresholds of | log2 fold-change | > 2 and FDR-adjusted P-values < 0.01.